
This page is dedicated to the Latent Heritable Confounder MR (LHC-MR) method. The paper can be found on medRxiv and the corresponding R-package here.
Our calculated LD scores can be downloaded here, whereas the spike-and-slab parameters of the local LD patterns for each SNP can be downloaded from here.
lhcMR is an R package that performs bi-directional causal estimation between a pair of traits, while accounting for the presence of a potential heritable confounder acting on the pair. The method termed LHC-MR aims to overcome some limitations seen in standard two-sample Mendelian Randomisation (MR) methods such as potential sample overlap, under-exploitation of genome-wide markers, and sensitivity to the presence of a heritable confounder of the exposure-outcome relationship.
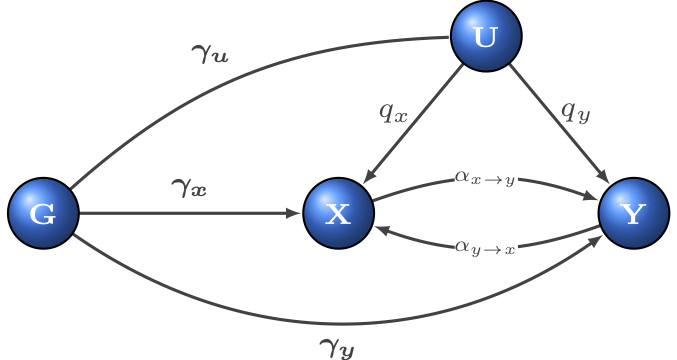
To do so, our approach builds on the typical MR framework to incorporate the presence of latent heritable confounder in a structural equation model (SEM). Given starting points for the parameters we wish to estimate (including bidirectional causal effect, confounder effects, direct heritabilities and more), LHC-MR optimizes the likelihood function to obtain maximum likelihood estimates for the parameters. LHC-MR then runs a block jackknife approach in order to calculate the standard error of the estimated parameters.