
10X Genomics Next GEM technology
GTF operates 10X Genomics solutions for high throughput single cell profiling.
We run both the standard Chromium and the newer Chromium X which required for the Single cell fixed RNA profiling application from 10X genomics (human and mouse only) and the High Throughput assay (twice more cells processed for roughly the same price).

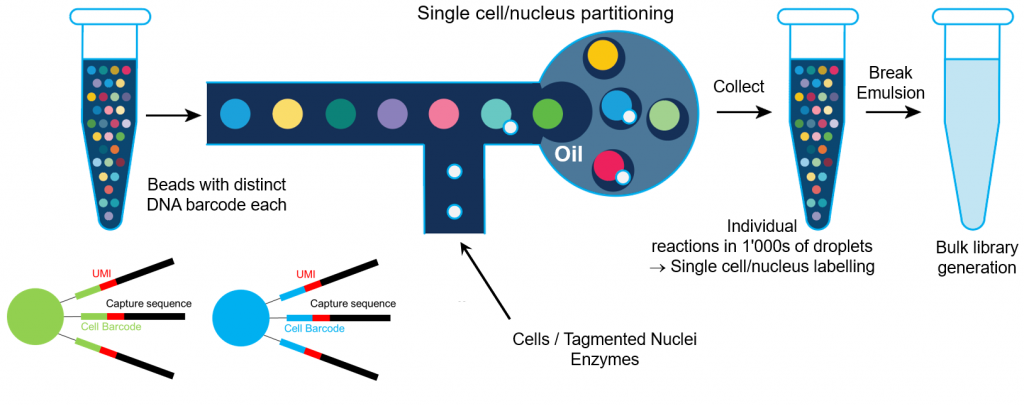
10X Genomics trick is based on single cell/nucleus emulsion. A DNA barcode, unique in each droplet, is linked to DNA/cDNA. The emulsion is broken and the subsequent sequencing library generation steps are performed bulk.
The standard gene expression assay is based on the generation of full length cDNA generation (primed at polyA tail) followed double strand cDNA amplification and generation of a 5′-end or 3′-end sequencing library generation. It requires cells are fresh and high viability. It works on any organism, provided the RNA is polyadenylated.
With the the fixed cells protocol, gene expression is achieved by sequencing RNA bound probes. Fixation with PFA leaves way more flexibility for sample preparation. Probes are however available only for human and mouse transcriptome. In addition, only gene expression applications are possible (not ATAC-Seq or VDJ sequencing).
Up to 10’000 cells/nuclei can be assayed in a single well, i.e. up to 80’000 cells per chip. However, the more cells or nuclei are loaded, the more multiplets may be present (see for instance this 10X Genomics note). In addition, more cells mean higher sequencing effort required (see below). A common target is in the range of 8’000 cells/nuclei per well.
With the high throughput assay, these numbers are doubled for roughly the same 10X processing cost. By using cell multiplexing (see below), it is possible to recover up to 30’000 cells per well. This is however bound to several limitations (please inquire).
Applications
Main 10X Genomics single cell applications are:
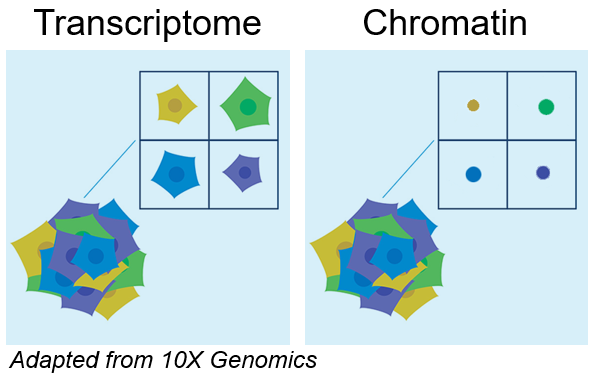
- Single cell transcriptomics
- Single cell epigenomics (chromatin) with ATAC-Seq
- Combine transcriptomics and ATAC-Seq (” multiome”)
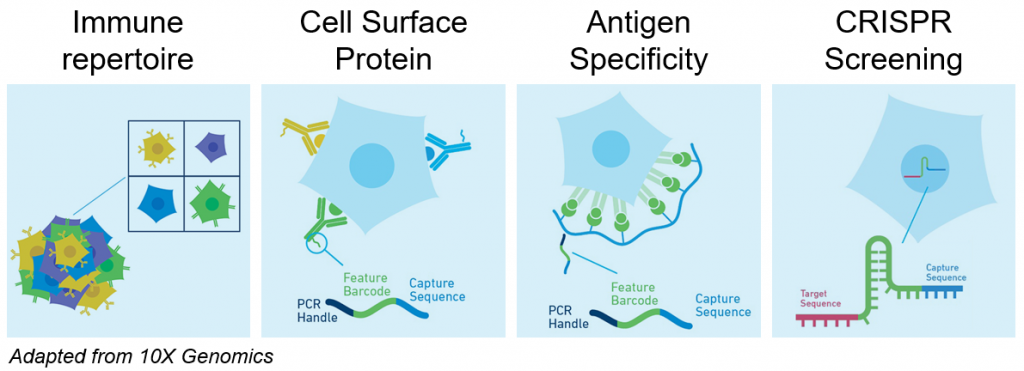
Single cell transcriptomics is not restricted to RNA profiling since each single cell analyzed can optionally be further characterized with:
- Immune repertoire profiling (Human or Mouse B or T cells V(D)J sequencing)
- CRISPR library sequencing for Perturb-Seq (also known as CRISP-Seq or CROP-Seq)
- Cell surface protein expression with DNA barcoded antibodies (also known as CITE-Seq or TotalSeq)
- Antigen Specificity with dCODE Dextramer
Schedule an experiment
Samples should be processed as quickly as possible after the cell sorting. Accordingly, experiments must be carefully scheduled with our staff. Please contact GTF as early as possible when designing your project.
In order to maximize cell viability, the 10X Chromium may be operated in one of the Lausanne University Cell Sorting facility (FCF, at Agora or Biopole) laboratory by a specialized GTF technician (service available upon request). This service is only available for the standard Chromium, not the Chromium X.
Cells or nuclei preparation
Cells or nuclei preparation is of user’s responsibility !
The cells/nuclei suspension quality is crucial for a successful single cell project. Cells/Nuclei must be perfectly dissociated, their density accurately quantified, and viability must be above 70% (for transcriptomics applications). Nuclei preparation is even more challenging since viabilitycheck does not apply (see 10X note).
Any manipulation (e.g. concentration by centrifugation) or waiting time can impact cells/nuclei quantity and quality. In particular, cell count and viability output metrics from cell sorters cannot be taken for granted. It is more than highly recommended that a final quality control step is performed on the very final material you provide us (disclaimer). You may want take advantage of our Moxi V cell counter which is optimal for several 10X Genomics applications (see this technote). We also operate the Luna-FX7 which can take advantage of AO/PI staining to accurately distinguish cells from debris.
Failure to provide good quality starting material will inevitably result in suboptimal, sometimes useless, results. Data quality can only be assessed after sequencing, meaning that expensive 10X and Illumina reagents are engaged, whatever the outcome is.
In any case, we strongly recommend that pilot experiments are performed in order to validate the cell preparation procedures.
The Chromium loading efficiency is in the range of 40% to 60%. This means that only ≃ 50% of the cells/nuclei loaded in the chip’s wells will actually be assayed. Given there is also generally around 50% yield for the cell preparation step (cell sorting and subsequent washings), we recommend starting the preparation procedure with at least 4-fold the amount of targeted cells/nuclei.
As a starting point, please refer to the following 10X genomics documents: Sample Preparation Tips for Single Cell Gene Expression (transcriptomics applications), Nuclei Isolation for Single Cell ATAC Sequencing (ATAC-Seq application), or Nuclei Isolation for Multiome (Gene Expression + ATAC seq from the same nuclei).
Cell or Nuclei Multiplexing
Samples may be multiplexed before being loading on the 10X Chromium chip.
The process is based on an extra DNA label added to the cells (or nuclei), or on barcoded probes when working with fixed cells (see below).
This saves reagents in case many samples have to be run in parallel, but decrease the number of individual cells assayed for each condition.
10X Genomics proposes an official solution with the CellPlex technology. So far, it is supported with the 3′-gene expression kits only.
Multiplexing can also be achieved with DNA barcoded antibodies as a process called Cell Hashing
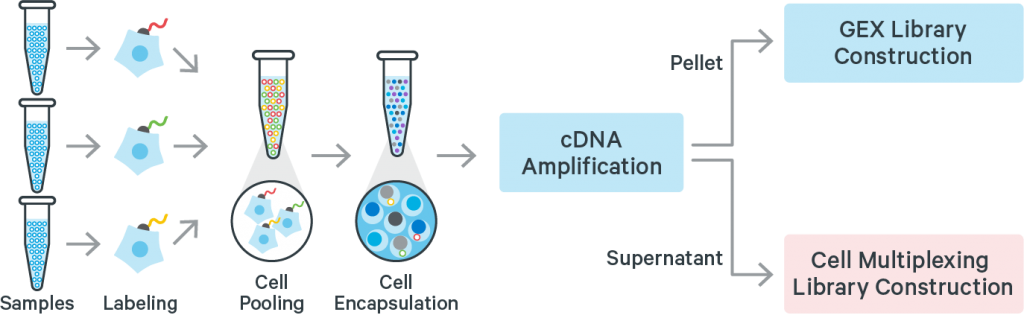
With the fixed cells kit, the multiplexing barcode is brought with the RNA detection probe. This solution is cleaner (less background) than CellPlex. It is also way more convenient since multiplexing can be designed a posteriori, after all cell handling experiment have been performed.
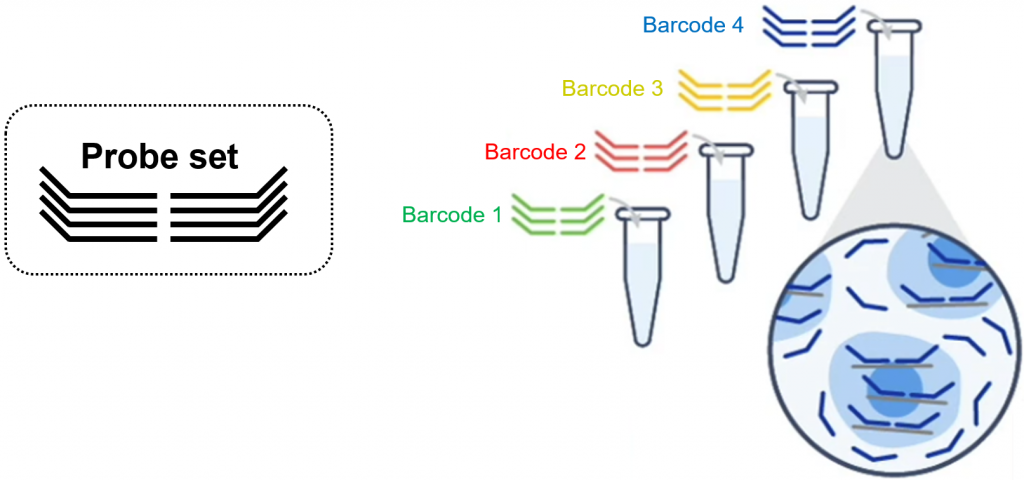
Setting up a multiplexing experiment is not trivial. In addition, it requires a large excess of cells since multiplex labelling requires many washes and results in cell loss. Please contact GTF for further information.
Spatial Transcriptomics
10X genomics offers Spatial Transcriptomics solutions.
It is available for fresh frozen samples, as well as FFPE material (but for human and mouse only).
Be aware the current resolution is pretty low, with 55 um wide spots.
Spatial transcriptomics involves multiple fields of expertise, not related to genomics, upstream of the sequencing library preparation step. Experimental procedures must be discussed in details with GTF, before planing any experiment.
Digital Spatial Profiling technology (DSP also known as GeoMx) from Nanostring is also available, but not operated by GTF. Please inquire for further information.
Library preparation
Once the user has prepared the cells/nuclei suspension, GTF will operate the 10X Genomics Chromium and generate the sequencing library. All 10X genomics reagents as well as advanced library quality control are included.
Chromium cell processing can optionally be split into several batches (i.e. chips) in order to avoid samples waiting too long. This must be discussed ahead with GTF.
Sequencing
We perform 10X library sequencing on the NovaSeq. The output is 400M filtered/usable reads per lane. The total amount of sequencing required is directly dependent on the number of cells loaded.
For ATACseq, the 10X Genomics recommendation is 25’000 reads per cell (Human or Mouse genomes).
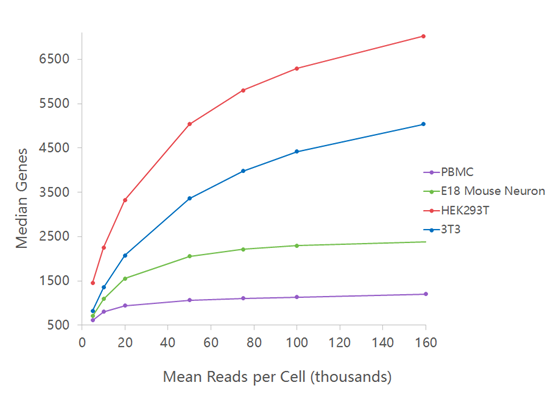
For RNAseq, 10X genomics recommends around 25’000 – 50’000 reads per cells but this number is greatly influenced by the complexity of the transcriptome (see figure below as well as this Technical Note). In addition, special interest in low expressed genes (e.g. transcription factors or long non coding RNA) may impose a higher sequencing depth.
A good practice is to request a single sequencing lane to start with. Further sequencing can then be requested after quality control for validating cell number, evaluating transcription saturation, and inspecting the presence of some genes of interest.
For Spatial transcriptomics the recommendation is 50’000 reads (fresh frozen material) or 25’000 reads (FFPE material) per spot. A capture area is made of 5’000 spots but only partially covered by the tissue slice. Sequencing depth requirement can be calculated by estimating the percentage of capture area occupancy.
Data analysis
GTF 10X Genomics service includes primary analysis with Cell Ranger software using standard settings. Deliverables are fastq files as well as the corresponding Cell Ranger count file outputs.
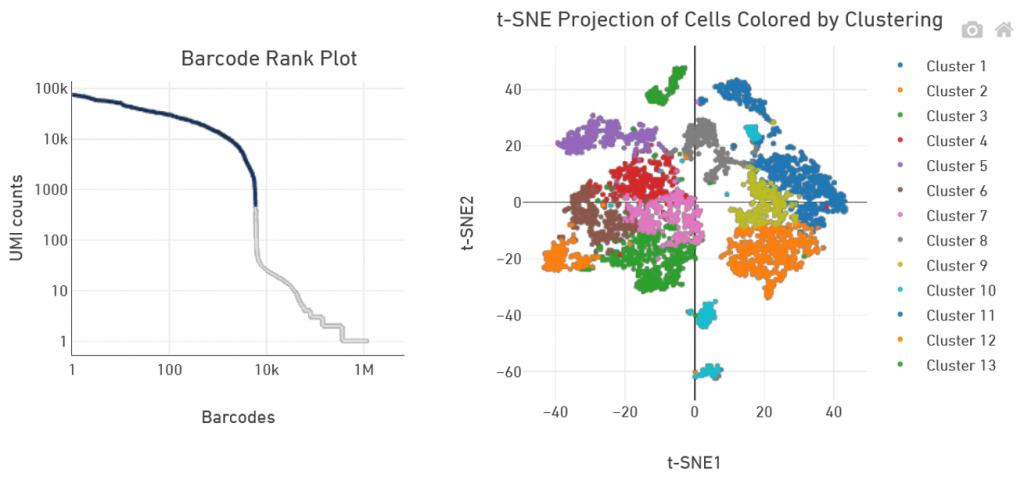
Secondary analysis, with refined clustering or pathway analysis for instance, is not proposed by GTF.