
See input guidelines at the bottom of this page
Service offered
The GTF team is in charge of performing library preparation and sequencing. The starting material (genomic DNA, RNA, or PCR amplicon) must be prepared by the user.
For long read sequencing, our standard analysis service is restricted to quality control and delivery of the raw data (demultiplexed when applicable). Some more advanced analysis supported by the Pacific Biosciences “SMRT Analysis” pipelines can be performed upon request.
Long read sequencing applications generally require more fine-tuning than standard Illumina Sequencing. We strongly recommend you discuss your project with us before starting experiments (in particular genomic DNA extraction).
Typical sequencing effort requirement
Official recommendations from Pacific Biosciences are shown in the table below.
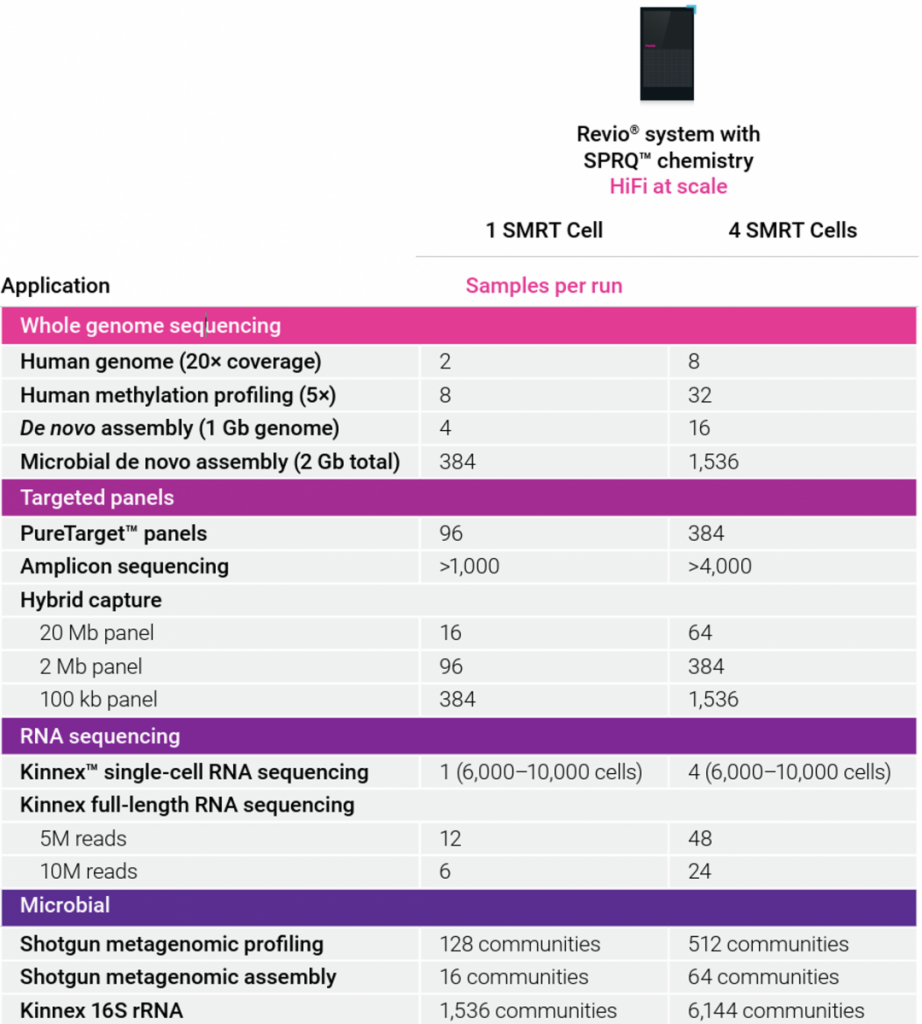
These numbers are to be fine tuned depending on the application. For genome sequencing for instance, some parameters such as genome complexity (repeated elements), ploidy, or level of heterozygosity, will impact the actual required sequencing effort.
For human genome sequencing, some more precise numbers are proposed:
Multiplexing strategies
HiFi sequencing is compatible with multiplexing. It can be low-plexing for demanding applications, to up to several 100s for amplicon analysis.
When working with non-amplified material (e.g. genomic DNA), multiplexing index (or barcodes) are added with the adapters during the library construction step. A collection of 96 barcodes is readily available.
If DNA template is obtained by PCR amplification, barcodes can be brought directly with the primers. All sample amplicons can then be pooled and a single library is generated. Depending on the project constraints (see pros and cons in the short reads amplicon sequencing section), a 1-step or 2-steps PCR may be preferable. Utilization of barcoded adaptors with “native” (non indexed) PCR product is sometimes a preferred solution.
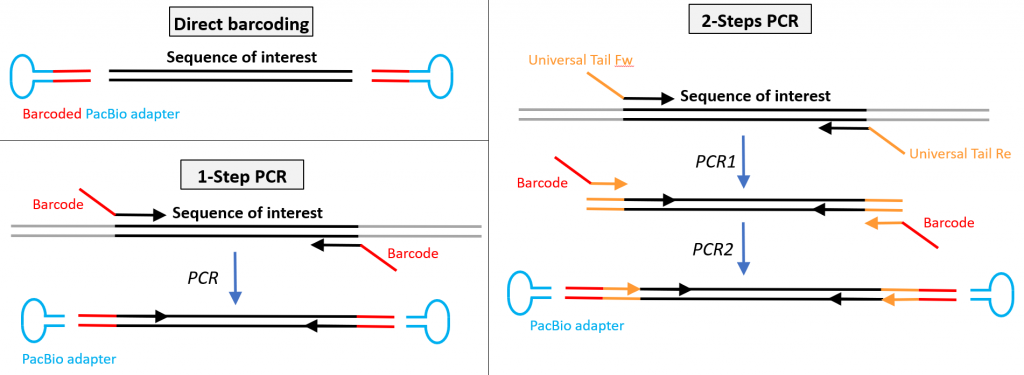
Full length bacterial 16S sequencing
GTF can provide barcoded PCR primers for “full length” 16S sequencing
Fw: 5’-GCATC/barcode/AGRGTTYGATYMTGGCTCAG-3’
Re: 5’-GCATC/barcode/RGYTACCTTGTTACGACTT-3’
- The workflow proposes is based on this 16S PacBio supported procedure
- Multiplexing relies on a “1-Step PCR” strategy (see above)
- PCR primers are provided pre-mixed (Fw + Re) and arrayed in full 96-wells plates”
- 96 combinatorial barcodes are available
- Users are in charge of performing the 16S PCR amplification (around 1.5 kb product)
GTF can pool and purify PCR products (optional), proceed to SMRTbell library preparation, and perform Revio sequencing
Typically, a single SMRT cell is sufficient for 96 samples (depends upon populations’ complexity)
Genomic DNA input quality
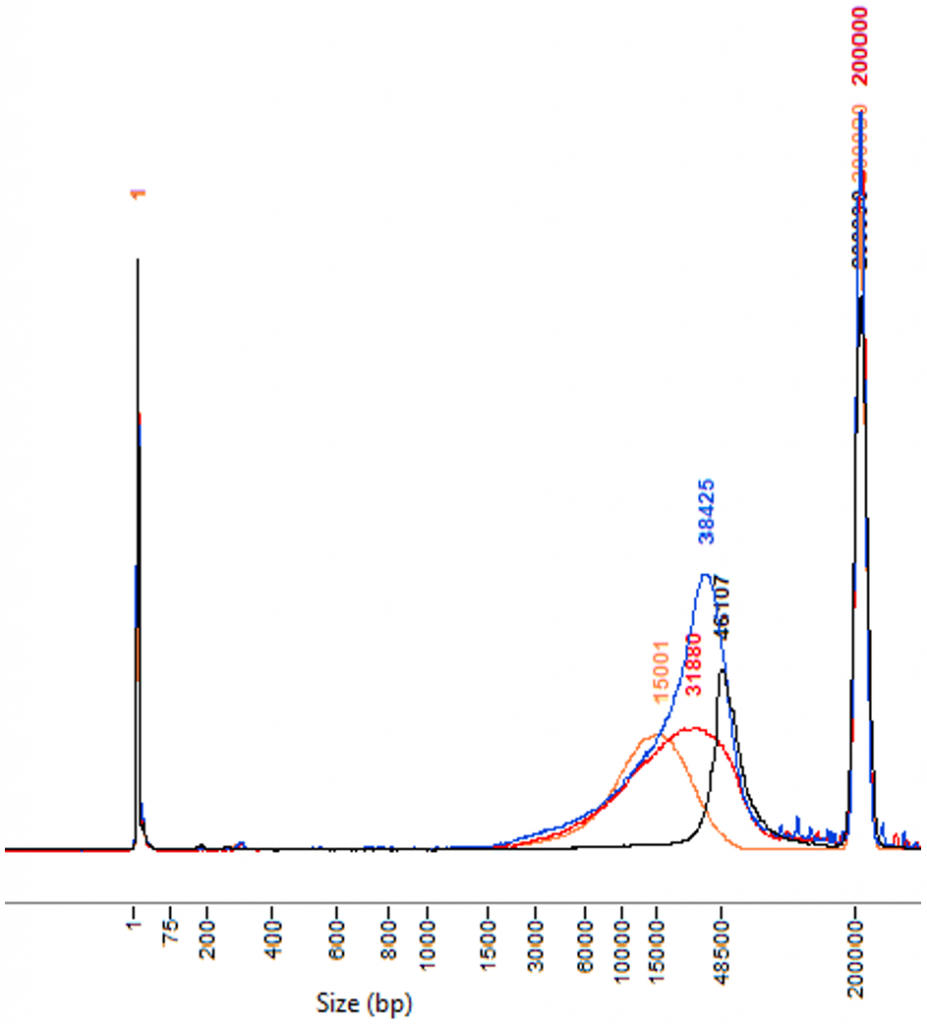
Starting material must be high molecular weight and high purity, without much lower size material present.
The presence of a lower molecular weight tail is generally a sign that DNA quality is suboptimal. As part of the library preparation procedure, DNA input will be sheared under controlled conditions. We repeatedly observed that suboptimal quality DNA get sheared to lower average size than good quality DNA. As a consequence, the amount of remaining material within the expected 15 kb to 25 kb size window can get too low for being processed into a sequencing library.
Below are typical input DNA size patterns obtained with the Fragment Analyzer
SMRT sequencing is sensitive to some contaminants which may be co-purified with DNA (e.g. sugars). Some contaminants inhibit the sequencing polymerase and drastically reduce sequencing yield. Most of DNA sources are fine but some organisms can be problematic. There is unfortunately no procedure to evaluate that (rare) issue prior to sequencing.
Genomic DNA input quantity.
Standard input HiFi sequencing requires at least 1 ug genomic DNA. Higher amount allows a more stringent size and quality selection of the final library and helps maximizing the final sequencing yield. More material is necessary if several SMRT cells are required.
Multiplexing several samples, from as low as 300 ng each, is a possibility when less material is available. Because less library can be generated for each sample, this limits the sequencing yield and therefore the size of the genome to be analyzed.
Ultra-low input requires 1 to 50 ng input DNA. The library preparation protocol includes a PCR amplification step.
Support genomes up to 3Gb
When too low amount of material is available, it is possible to pool several individuals to reach the targeted DNA input amount. Practically, since HiFi genome sequencing is haplotype aware, it is required that the heterozygosity level is low (ideally clonal individuals).
GTF input material guidelines
In this document are the sample preparation guidelines proposed by GTF (based on official recommendations from Pacific Biosciences).
Pacific Biosciences offers extraction kits with sample-specific protocols for a wide range of specimen types.