|
|

|
|
|
|
|
Facility News
We did it ! It is our first Newsletter Anniversary 🎉&💃 ! Thanks for being supportive, giving us feedback and showing in your experiments that our teachings are helping you. Truly a blessing to have such a nice group of users !
|
In this month FACS Tips will give you some more insights into what makes a efficient cell staining and how you can improve the quality of your data as a consequence.
|
This month's great news is that we will soon have a new instrument in demo at the Epalinges site. The Attune CytPix from ThermoFisher has the ability to deliver brightfield images at the same time as fluorescent parameter like a conventional flow cytometer. Additionnally, the machine can run whole blood without lysis at a fast pace and still provide great results. The machine will be available for FREE from June 1st to July 8th and you can start making reservations starting May 16th on IRIS. So please feel free to use it and give us your impressions.
|
|
For the Newsletter Quiz, congratulations to Chakradhar Yakkala from the Kandalaft Lab ! This month's answer were (12 channels, 642 nm, False) and the amount of ImageStream hours were 171,25 hours !
|
|

|
For the May Quiz, we a trying something a bit different to make more accessible and a bit more eco-friendly. You simply have to click on the image at the end of the newsletter to access an online form to answer !
|
|
|
|
FACS Tips
What Goes Into (And Comes Out Of) Cell Staining
Sometimes it's the simplest things that can be our downfall in experiments, and this is especially true in staining. Many little things have to come together properly for us to have high quality samples to create great data. By overlooking steps or cutting corners on our staining protocols we risk undoing all the hard work that goes into each experiment. It’s important to think about why certain things are done and why the process is the way it is. This can help us move through our staining process with the goal of minimizing cell death and debris, while bringing out the best quality specific staining.
|
Buffers
Starting from the basics, the buffer itself. The flow cytometry staining buffer, as we call it FACS buffer, is most commonly a PBS or HBSS solution, while both are isotonic solutions they each have preferred uses. PBS is a buffered salt solution and is better at managing pH, while HBSS is a balanced salt solution containing glucose which can better maintain cell viability during longer protocols. It is also beneficial also to include a protein like BSA (0.5-1%) or FBS (2-5%) in the FACS buffer, this is because proteins are unspecifically adsorbed on/in cells at various levels. If the staining or washing buffer lacks proteins, then antibodies have a greater likelihood to bind non-specifically to cells creating higher background autofluorescence. These added proteins reduce nonspecific heterophilic binding of antibodies while preserving cell viability. Once added protein concentrations go beyond these typical values for our FACS buffers you can expect disruption in antibody binding as well as more autofluorescence. It’s important to note that if you’re preparing your cells for sorting instead of use on the analyzers, a lower concentration of protein is advisable in your FACS buffer, ideally <2% FBS />
|
For staining panels involving several polymer dyes (BUV395, BUV737, BV605, BV711, and SuperBright dyes), both BD and ThermoFisher have developed special staining buffers to overcome problems with the polymers interaction with PBS buffer causing aggregation and overestimation of double positives and increased background in some channels of the violet laser. By using the Brilliant Staining Buffer (BD Biosciences, https://www.bdbiosciences.com/en-ca/products/reagents/flow-cytometry-reagents/clinical-diagnostics/buffers-and-supporting-reagents-ivd-ce-ivd/brilliant-stain-buffer.659611) or Super Bright Complete Staining Buffer (ThermoFisher, https://www.thermofisher.com/order/catalog/product/SB-4401-75) this should be prevented.
|
If you’ve ever wondered why you can’t stain in media it is due to the buffering system it maintains for CO2-carbonate. Media is designed to be used at high atmospheric CO2 (5%), however at typical atmospheric CO2 (0.04%) the CO2 in the media evaporates, driving an increase in pH, which may eventually cause cell death. Additionally, phenol red is highly fluorescent when excited at 440 nm, and it can increase background during analysis.
|
Clogs can be a particularly large problem in our experiments, when we’re running 20 or 30 samples in a single session a clog on the machine can slow us down and even potentially ruin our experiment. If you know you have particularly adherent cells or considerable cell death causing DNA release it is essential that you include EDTA (0.5-5mM) in your FACS buffer. EDTA conceals free Ca2+ and Mg2+ ions in solution which inhibits calcium dependent cell adhesion molecule and integrin interactions. DNase (25-200ug/mL) is also useful in the reduction of cell aggregation due to free DNA in solution, however it should be considered that when used in combination with EDTA, which actively chelates Mg2+ ions, the presence of EDTA will reduce the effectiveness of the DNase. Rather EGTA should be used in combination with DNase.
|
Temperature
|
Most often we stain at 4 C, this helps maintain cell viability and also reduces the chance of internalization of surface markers. It’s important to pay attention to the manufacturer's instructions though, not all antibodies are added at 4 C. One notable exception is CCR7 which stains best at 37 C. Chemokine receptor detection is another area where staining at 4 C may not be best and adjustments will need to be made. In addition to temperature, it's important that samples be left to incubate without light to avoid photobleaching of the fluorophores, in fact concealing samples from light all the way up to the time they are run on the machine will best preserve their functionality. Incubation time for staining is typically 20 minutes, there are some exceptions though. For some streptavidin conjugates, PerCp-Cy5.5 and PE-Cy5 for example, time should be minimized to 15 mins maximum or the background increases dramatically. So again it’s important to pay attention to your particular staining panel.
|
|
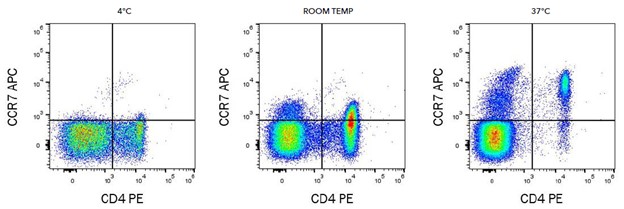
|
Image from bio-techne
|
Specific vs Non-Specific Staining
|
Antibodies can bind to cells in a variety of ways, the problem is that we are only interested in antigen specific binding. Antibody binding to an exposed Fc receptor on the cell, or non-specifically, by ionic or hydrophobic interactions between the antibody and the cell can cause false positive staining and needs to be addressed in our staining protocol. The goal of blocking is to prevent antibodies binding with an Fc receptor on the cell, such as CD16, CD32, and/or CD64. While many cell types can express these receptors, samples containing monocytes, Dendritic cells, NK cells, and/or neutrophils can be especially high in Fc receptors and the need to block samples becomes more necessary. Blocking works by adding specific blocking antibodies prior to staining to sequester all the available Fc receptors in the sample. You need to block with the IgG of the species whose cells you're staining, for mouse tissues, block with mouse IgG; for rat tissues, block with rat IgG. Miltenyi has developed recombinant antibodies (REAfinity) that have been engineered to produce highly specific antibodies that require no FcR blocking step.
|
|
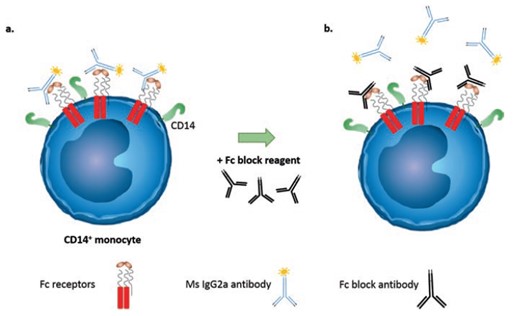
|
Image from Goetz, C., Hammerbeck, C., & Bonnevier, J. (2018). Flow Cytometry Basics for the Non-Expert [electronic resource] (1st ed. 2018.). Cham: Springer International Publishing. https://doi.org/10.1007/978-3-319-98071-3
|
Washing is an essential step following staining to ensure any unbound antibody can be removed. By increasing the number of washes it should improve the resolution of the positive stain from the background, however, too many washes and you can start to lose cells or influence viability. Two or three washes would be recommended after staining as well as in between steps in your staining protocol if you are working in small volume containers like 96-wells plates. One advantage of staining in 5ml tubes is you can wash in a larger volume, which should also improve the removal of unbound antibody and debris compared to staining in plates, were wash volume is much more limited.
|
Conclusion
|
Everyone can have slightly different protocols when it comes to staining, with subtle changes to the buffers, incubation times, and washing steps. What’s important is that it works for you and your experiment to get the best results. This is certainly not an exhaustive list of advice for staining optimization, and most of the advice provided here is geared towards surface staining rather than intracellular. There are always opportunities to improve your staining protocol and we’re always happy to offer support and advice if you think you would like to make some changes.
|
|
|
|
Can you answer this ?
|

|
|
|
|