|
|

|
|
|
|
|
|
|
Facility News
We're soon hitting mid-July and you know what it means, a well deserved break for all ! So as you are enjoying your holidays ☀️ and take a break from science, don't forget your favorite FCF team and feel free to share pictures of your travels and explorations with us ! Especially if you're hitting one of those coolcation spots so trendy at the moment (looking at you, Stockholm).
|
In this month FACS Tips, Kevin is talking about an interesting technique that was sometimes used back in my flow days, stacking ! Read all about it now !
|
|
Stanislav Dergun won the mug of the month below, congratulations to you ! Please come by to our office at the SE-C Biopole to pick it up !
|
|

|
Please try to answer our quiz it and get a chance to win our next mug of the month ! Good luck to you all !
|
|
|
|
|
|
FACS Tips
|
How far can we stretch a single fluorophore?
|
There's a simple philosophy in flow cytometry: one marker, one fluorophore. That's how we best discriminate things. We design our panel such that we know BV421 is CD4, that APC is Ly6G, that RB705 is CD19.
|
Increasingly, the field is moving toward a place where 40+ colour panels are the priority. We want to expand the information we capture as much as possible by building bigger, bolder panels, and here at the UNIL FCF we have several instruments that can do exactly that, between our Aurora and S8 spectral platforms.
|
But that got me thinking about the opposite situation. What does it look like if we try to cram as much information as possible into as few fluorophores as we can? What happens when we reuse a fluorophore for multiple markers, not just as a dump channel, but for genuine markers of interest?
|
|
Turns out this is a real technique, and it even has a name: stacking. It relies on a deceptively simple idea, a fluorophore can be repeated as long as you can be certain the markers sharing it sit on mutually exclusive cell populations. The cytometer cannot tell which antibody contributed a given photon, so stacking only works when the biological logic of the experiment guarantees that no single cell will ever co-express the markers sharing that channel. You then rely on the gating strategy to resolve which marker the signal represents by context: you already know which lineage you're looking at before you interrogate that channel.
|
|

|
|
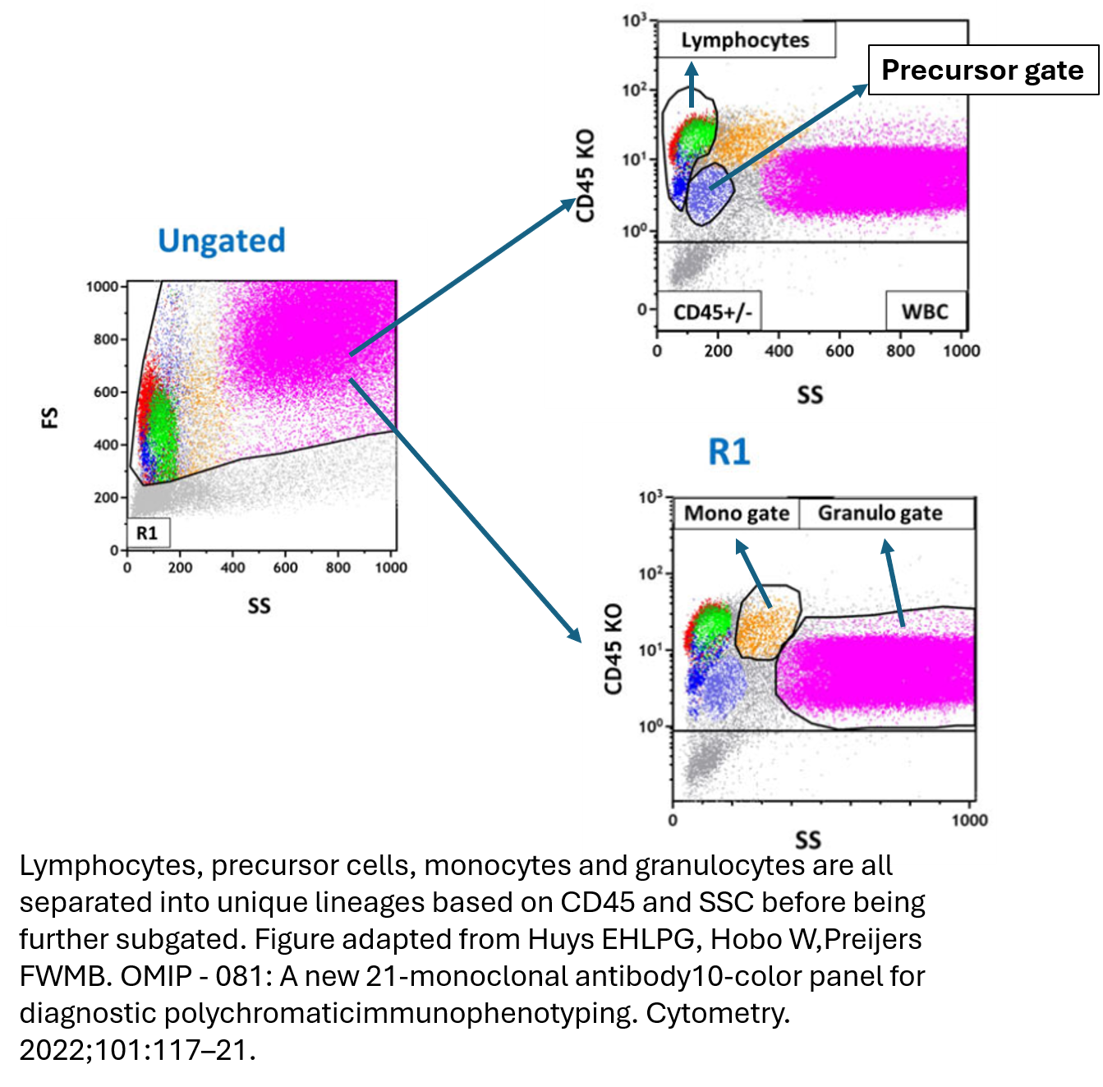
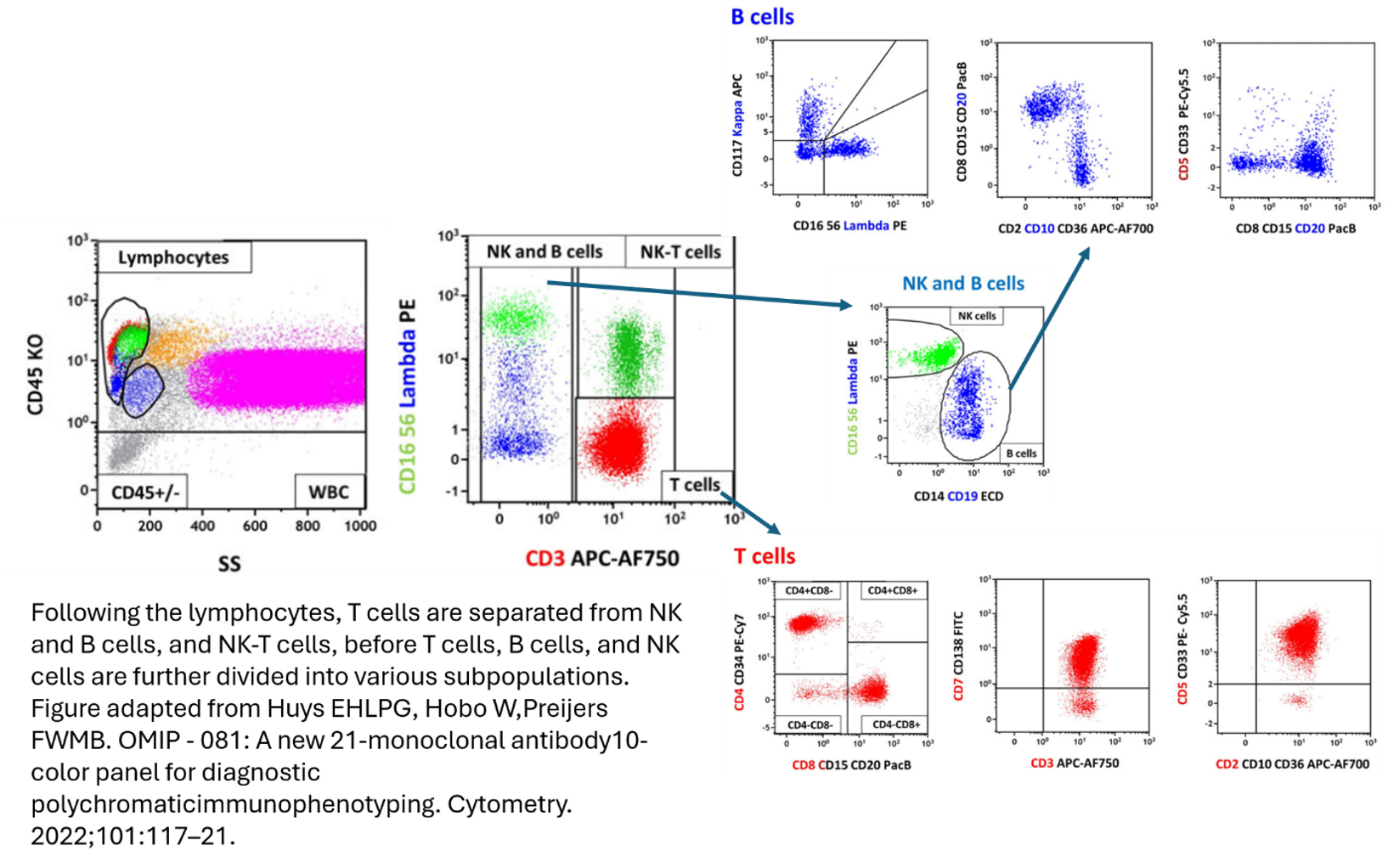
There are some nice examples of this in practice. In OMIP-081 (A new 21-monoclonal antibody 10-color panel for diagnostic polychromatic immunophenotyping, Huys et al.), stacking is used to cleverly resolve multiple cell types that share overlapping fluorochromes.
|
|
|
|
|
In this panel, demonstrated in the figures above, they’ve used a highly involved gating process to separate out cell lineages, with FSC/SSC properties, and pan lineage markers, to then begin to individually phenotype subsets with overlapping markers depending on the lineage. For example, Pacific Blue is used to stain both CD8 and CD20, however, the T cells and B cells are already separated upstream so both can still markers can still be effectively separated once in their individual gating stream.
|
|
|
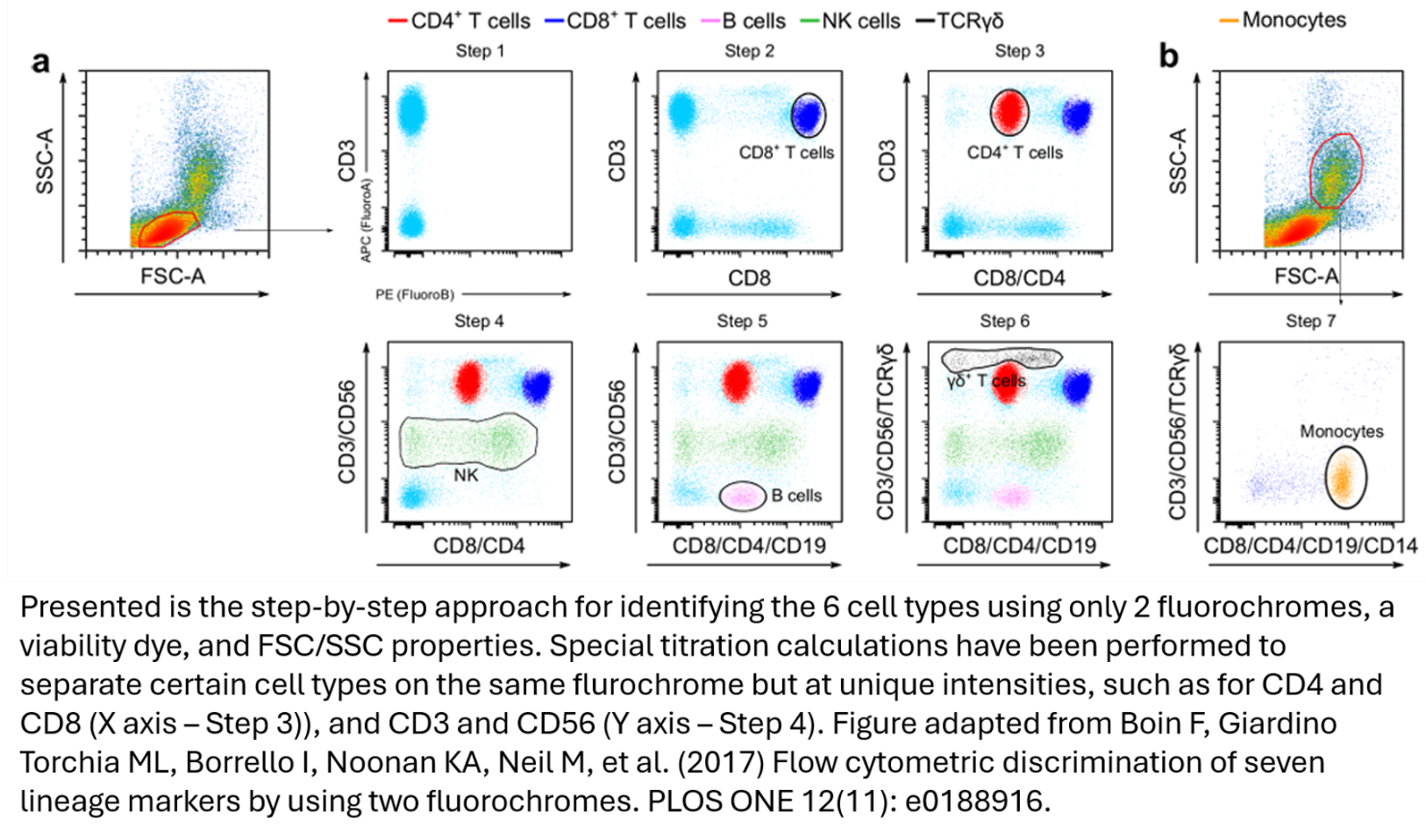
An even more aggressive version of this strategy appears in the paper Flow cytometric discrimination of seven lineage markers by using two fluorochromes (Boein et al.). Their strategy, just two fluorochromes plus a dead-cell dye, combined with careful subgating on FSC × SSC, reliably discriminates CD4+ and CD8+ T cells, γδ T cells, B cells, NK cells, and monocytes. They don't just overlap fluorochromes; they use carefully managed titrations to separate populations by dim versus bright expression within the same channel, sometimes on both the X and Y axes at once. For example, CD4 and CD8 are stained with the same fluorophore, but titrated such that the CD4 cells sit at half the intensity value of the CD8 cells.
|
It's an impressive act of flow cytometric contortion, that many assessments squeezed out of such a limited experiment. They go on to suggest it could serve as a useful backbone to add more colours to in future work.
|
Typically, though, this isn't something we'd advise in the FCF. There are many places it can create problems. For one, marker expression can be complicated, and you have to be very sure about what you're stacking. Take CD4, a T cell marker, it's also expressed at low levels on monocytes. Or CD8, found at low levels on some NK cells. CD56 and CD16 both turn up on T cells and monocytes as well. The "mutually exclusive" assumption is doing a lot of work, and biology doesn't always cooperate.
|
Beyond the difficulty of guaranteeing exclusivity, there's also non-specific background, which can be additive. It becomes essential that the antibodies are properly balanced: if too much of one is used, its non-specific staining can outweigh the specific staining of another antibody in the same stacked channel if that one is dim. This is no longer a simple titration of each antibody on its own, but a comparative one that still has to resolve every fluorochrome involved.
|
As it stands, we have plenty of modern instruments capable of reading enough unique fluorophores that it's rarely worth playing a game of who-expresses-what and risking getting it wrong. These techniques were far more necessary in the pre-spectral days, when detector space was limited and the pressure to get more out of precious samples was higher. That's exactly the reasoning given in the OMIP-081 paper, which notes that clinical cytometers have historically been more limited in detector space, as spectral technology has been adopted more slowly on the clinical side.
|
|
So stacking sits in an interesting spot: a clever, still-useful tool when constraints demand it, but one that asks you to trust your biology completely, and on today's spectral instruments, that's usually a trade we don't need to make. If you’re curious how stacking could be a useful tool in your experiments, come discuss with us at the FCF.
|
|
|
|
|