|
|

|
|
|
|
|
|
|
Facility News
Happy (late) Easter to you all 🐰🥚 ! I hope you could enjoy the first real break of the year and the wonderful weather that Switzerland was blessed with for the past few days. I'm certain you were part of many chocolate eggs hunts in the forests and gardens of your families ! Like every year and to make it last a big longer, I will offer our users some chocolate eggs to grab at each of the facilities. This time around, they are not hidden, the are placed at the entrance of our labs so come by, say Hi and get your fill of chocolate 🍫 !
|
The big news of the month is the arrival of Naoill Abdelaoui in our facility 🎉 ! We're very pleased to see her join the Biopole SE-C team ! Naoill was previously working as the Imaging and Mass Cytometry Platform Manager of the Harari Group at the Agora. She has extensive experience in mass cytometry, cell sorting and complex data interpretation. Her expertise in batch effect correction will be of particular interest to some of the FCF users.
|
This month, Kevin is bringing to you a refresher about single stains in spectral flow cytometry and covers the Do's and Don't s to make sure you can get the best unmixing possible ! Please check it out !
|
|
Ana Belén Plata Gomez won the mug of the month below, congratulations to you ! Please come by to our office at the SE-C Biopole to pick it up !
|
|

|
Please try to answer our quiz it and get a chance to win our next mug of the month ! Good luck to you all !
|
|
|
|
FACS Tips
|
|
|
Single Stains: Pains or Gains
|
When our unmixing is wrong the first recommendation is usually to remake your single stains. That isn’t nearly as simple as it sounds though as we’ve discovered.
|
|
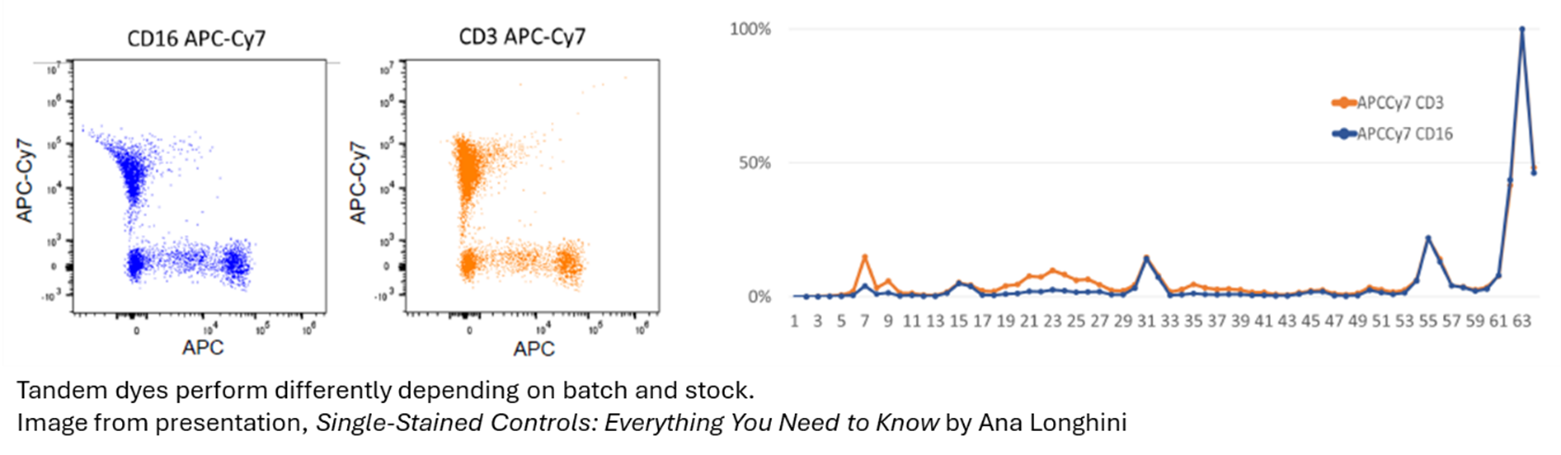
Despite our attempts, coming up with hard and fast rules for single stains for spectral cytometry has proven to be very tough. The variability is considerably greater than in conventional cytometry, largely stemming from tandem fluorochromes having inconsistent emission properties between batches. Fluorochromes can have instability from one sample to the next, they also perform differently depending on how they were treated, fix/perm or other. This can become complicated when matching single stains to samples.
|
|
|
On top of this, there are differences between instruments. Thankfully, we are predominantly using Aurora spectral machines, but even then we can anticipate some variation when trying to expand across multiple machines.
|
And even when we try to stabilize this all by using compensation beads, which ones? There are many different brands available and they don’t always perform well for spectral unmixing as we’ve discussed previously.
|
Although we may not be able to come up with perfect solutions if we can at least better identify where issues come from we can plan to limit them going forward.
|
With that in mind, let's approach these challenges by focusing on the three rules of compensation, a framework that helps us pinpoint exactly where things can go wrong.
|
Rule 1: Single stain colour and experimental fluorochromes must be the same
While this is a rule that gets broken somewhat frequently without much consequence in conventional flow, it becomes a crucial detail that can sink your experiment if not followed. By now I think we can all appreciate that APC can not be used for AF647 or FITC can not be used as a single for GFP.
|
But even when we do match our fluorochrome, the subtle and unique differences of how the fluorochrome behaves when it interacts with our cells, buffers, or beads can create subtle mismatches that suddenly make us break this rule without even realizing. So what do we do?
|
|
|
Unfortunately, our best recommendation currently would be to try multiple options and see what works best. If you’re working with complex tumor samples you can try single stains in that tissue type, but also in more simple splenocytes could work better due to less autofluorescence complication. If using cells, it would definitely be recommended to match the buffers and treatments between your fully stained and single stained controls. So if you use a brilliant staining buffer, monocyte blocking buffer, or a fix/perm reagent, you should also treat your single stains with it, that includes single stains in beads.
|
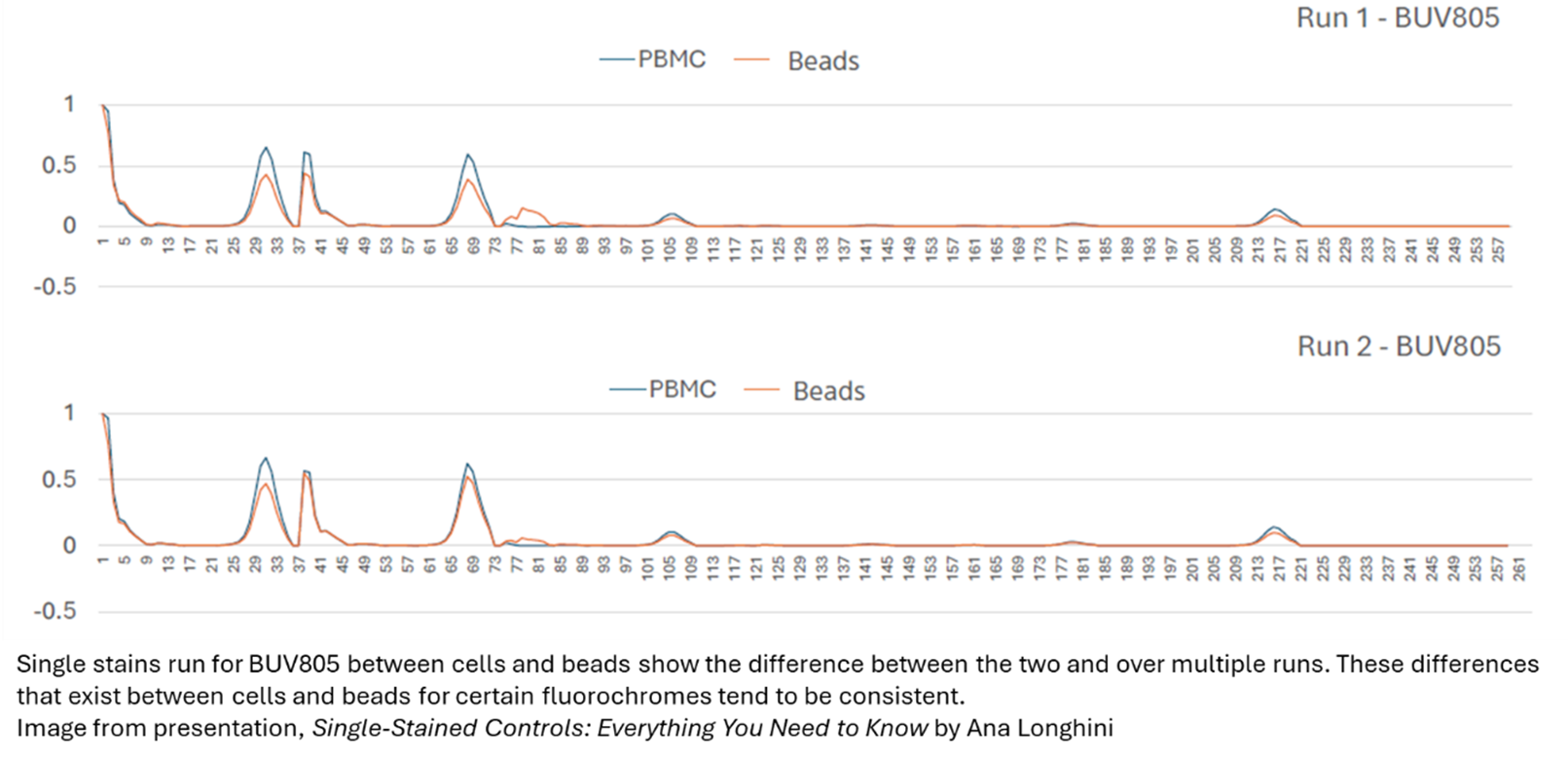
Also, anytime you change tandem fluorochrome lots, that is a good indicator that it is time to run a new single stain for that fluorochrome to reflect any changes that may occur from lot to lot, even of the same companies’ product. This is a common cause of unmixing error that can simply be fixed by making sure all tandem lots are up to date with their matching single stain.
|
|
|
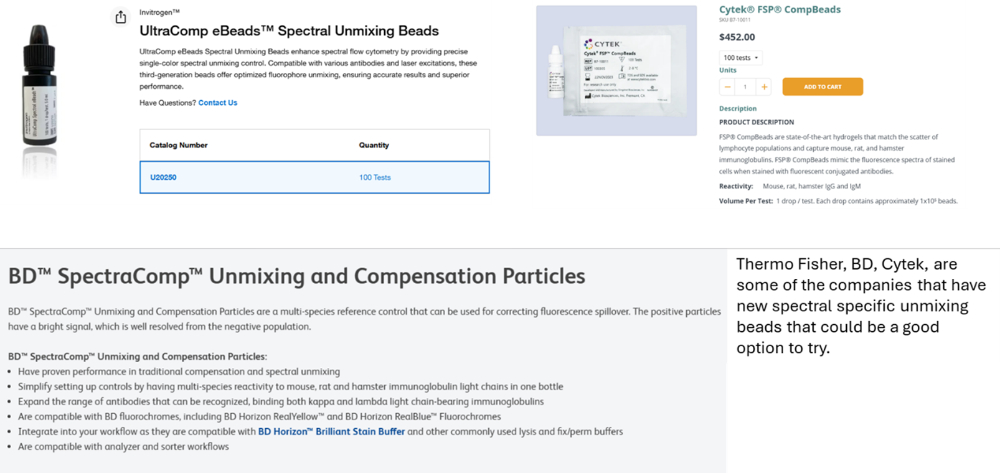
For beads, companies such as Thermo Fisher, BD, and Cytek are releasing new and optimized beads for spectral cytometry that are certainly worth exploring. They may not be perfect, but for some very hard to capture markers, beads may be the only option. In that case, it's best to pair these markers with non-tandem dyes to improve consistency.
|
As a last resort, for very rare hard to capture markers, it would be preferable to unmix with a single stain using a more plentiful replacement marker such as CD4 in a fluorophore like FITC or APC that has much more consistency and reliability compared to APC-Cy7.
|
Rule 2: The single colour controls need to be as bright or brighter than experimental samples
|
|
Again, beads tend to be superior in this situation as they non-specifically bind our fluorophores in a manner that tends to make them quite bright compared to cells. And if you’re having performance issues in your single stains in cells routinely being too dim compared to your fully stained samples, this could be an indication to switch to beads. Again, it is necessary to try both and see what performs best. We covered this topic in a previous newsletter (here)
|
|
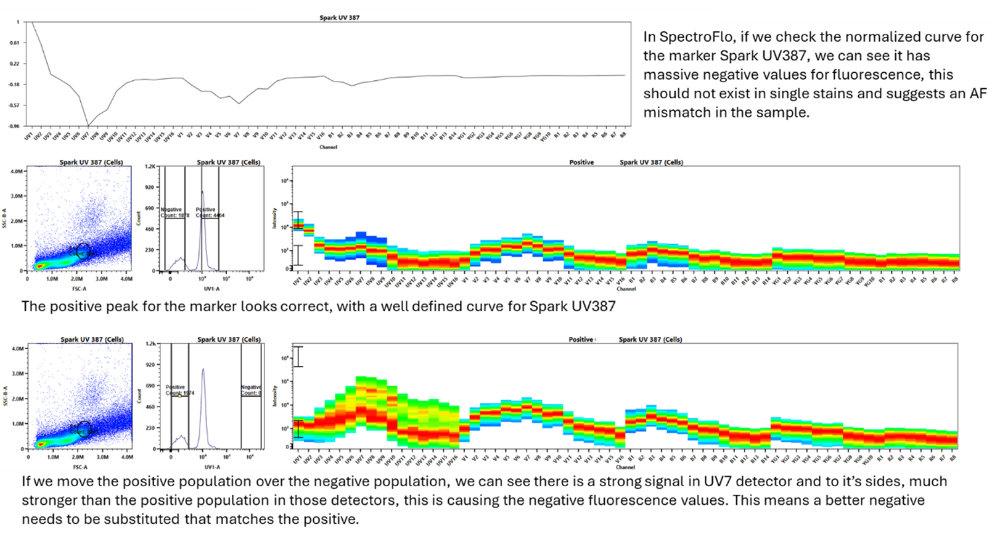
Rule 3: Autofluorescence of the positive and negative need to match
|
|
|
An easy way to see if your single stains have followed this rule in SpectroFlo is to look at the normalized single stain plots as the last step in the unmixing wizard. If you have a negative value it means there is likely an autofluorescence issue between the positive and negative population. This can be solved by more carefully selecting your positive and negative cells within the FSCxSSC plot ensuring you are only grabbing the cells of interest, or by using a universal negative or another more carefully planned negative that matches the autofluorescence of that specific cell type.
|
|
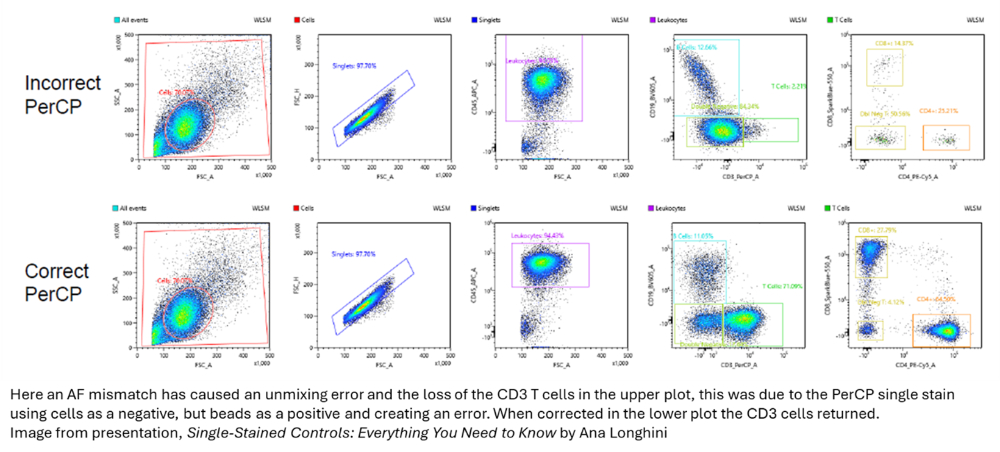
|
Beads have an advantage here as well, since the autofluorescence should be consistent between the positive and negative populations. It's also straightforward to prepare one unstained tube of beads as a universal negative for all bead tubes.
|
Conclusions
|
While beads would be much easier for many reasons, they don't always perform as needed for single stains. With cells, we enter a game of matching that can be challenging to get right. Things are always advancing, and these problems could well be solved with improved spectral beads and better protocols, but for the moment, the best we can do is identify where our problems come from and work to minimize them. Accepting imperfect unmixing may sometimes be necessary, and that's when we lean on panel design and gating controls like FMO to guide decisions when data appears imperfect. If you need help with your experimental single stains, feel free to reach out to the FCF staff, we're happy to help you optimize.
|
|
|
|
|