The study of the
Ursus lineage, including brown bear (
Ursus arctos), black bear (
Ursus americanus) and polar bear (
Ursus maritimus), provides the ability of addressing the subject of adaptation to extreme (salty and glacial) environments in mammals. Moreover, in last few decades, polar bears won public and media attention, being one of the most charismatic species endangered by global warming and Arctic ice melting. To trace history of innovations and determine response to environmental changes in populations of polar bears, two articles published in Science and Proceedings of the National Academy of Sciences in April and June 2012 provide new data and insights to resolve this question.
The absence of fossil of polar bears dating before the late Pleistocene (circa 126 000 years ago) and mitochondrial data, suggesting that polar bear were very closely related to a group of brown bear living in Admiralty, Baranof and Chichagof (ABC) islands in Alaska, previously led to believe that polar bears recently emerged from brown bears. The consequences of this hypotheses would be :
- Polar bear underwent a very rapid and recent (less than 200 ky ago) adaptation to extreme environment (previously not seen in mammals)
- Brown bear is a paraphyletic taxon, as polar bear is the sister specie of the ABC bears (see Fig. 1)
 |
| Fig. 1: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
|
|
Phylogeny of bear lineage with mitochondrial DNA and Bayesian maximum clade credibility model
The blue box contains polar individuals coming from Svalbard and Alaska and an ancient sample 130ky to 110 ky old, the yellow box ABC individuals and the pink box other brown bear individuals. The outgroup is made of black bears individuals.
Nevertheless, both fossil data, as it can be incomplete, and mitochondrial data, as it sensitive to hybridization, are not sufficient to confirm this hypothesis. Thus the two publishing groups led in parallel projects aiming to collect nuclear data and test its agreement with mitochondrial data.
Hailer et al., in their work Nuclear Genomic Sequences Reveal that Polar Bears Are an Old and Distinct Bear Lineage published in Science, sequenced 9116 nucleotides from 14 independent introns in 45 individuals of black, brown and polar bears. Introns were sequenced to provide more variation between individuals: given the low amount of time since the divergence of the last common ancestor of bears (estimated between 559 to 1 429 ky ago in their study), choosing exons, whose evolution being more likely bounded by selection, would have yielded less information.
Using this data and various phylogenetic reconstructions (bayesian multilocus coalescent approach, bayesian inference for the concatenated data and neighbour-joining of the differentiation estimates between species) that all led to the same conclusion, they recovered the three species of bears as being monophyletic and observed in the species tree the polar bear clade being sister to the brown bear clade. They estimated the divergence time of the two species around 603 ky ago (338 to 934 ky being the 99% highest credibility range) and clearly revealed a discrepancy with the mitochondrial data.
The authors resolved this incongruence by stating that the most probable scenario was a divergence between polar and brown species 600 ky ago and an hybridization event between 111 to 166 ky ago between polar bears and ABC bears leading to the complete replacement of the former mtDNA by the latter. The opposite phenomenon (several and severe introgression events of polar bears mtDNA into brown bears leading to all extant mtDNA being of polar origin) is judged very unlikely by the authors given the extended range of distribution of the brown bear. The lack of finding of older fossil from polar bears was explained by their constantly changing living environment.
Despite the recent hybridization event, Hailer et al. found very few common nuclear haplotypes between polar and brown bears: out of the 35 polar and 79 brown haplotypes, only 6 of them were shared across both species. Nevertheless, we must bear in mind that given the relatively low amount of nuclear data analysed, those findings might not reflect the entire picture of polar and brown bears nuclear DNA ancestry.
In Polar and brown bear genomes reveal ancient admixture and demographics footprints of past climate change, published in PNAS by Miller et al., a genome-wide sequencing project was adopted to unravel the same problem. In this extensive study, the authors assembled a reference genome of a polar bear individual, deeply sequenced the genome of two ABC, one black and one non-ABC brown bear (GRZ). Finally, they produced low coverage data from 23 other polar bear individuals, one of them being an ancient specimen 110 to 130 ky old found in Svalbard.
Having aligned all reads from every samples to the polar bear genome reference, they identified 12 millions of what they called “SNPs” (even though they are dealing with three different species) and constructed the following phylogeny (Fig. 2).
 |
| Fig. 2: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
Phylogeny based on the matrix of distances of the 12 millions SNP and using a neighbour-joining algorithm (probably given the amount of data and computational time needed with more sophisticated algorithms)
We observe that, as in the previous paper, the nuclear data is not in agreement with the mitochondrial data. A scenario where polar bears emerged as a sister species of the brown species and later experienced a massive and unique event of mtDNA introgression from ABC bears (as the polar bear individuals form only one group in Fig. 1) is again strongly favoured. Regarding the ancient polar bear specimen, both trees inform us that it dates after the mtDNA introgression event and that the modern individuals living in Svalbard are actually more closely related to the modern individuals in Alaska than to the ancient one.
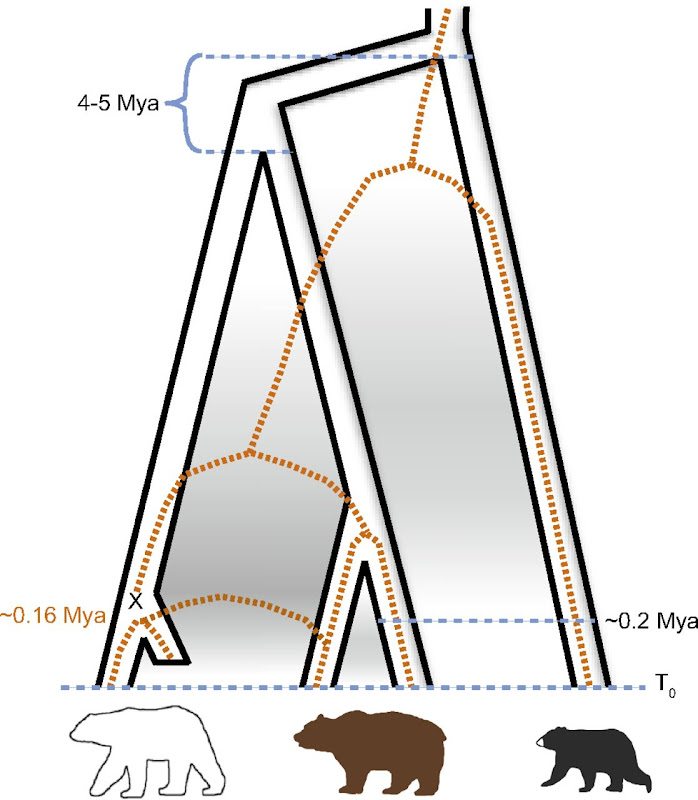
Though up to this point both articles seem consistent, following findings radically differ with the previous study. Indeed, Miller et al., used a coalescence hidden Markov model for four of their deeply-covered genomes (one ABC, one polar bear, one brown bear, one black bear) to assess the history of the lineage. They estimated both the splits of polar bears with brown bears and the common ancestor of those two species with black bears to have occurred around 4 to 5 My ago, as shown in Fig. 3.
 |
| Fig. 3: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
|
|
Reconstructed evolutionnary history of polar, brown and black bears
The black solid line represent the specie tree and the brown dashed lines the mtDNA tree
The X represents the introgression event, the shortened branch of the specie tree the disappearance of the ancient Svalbard lineage
It is however true that Hailer et al. reported on their article (that pre-dates the PNAS one) that other studies hint that the 600 ky-value is an underestimate of the splitting time of the two lineages under consideration, without it weakening their own conclusion.
Nevertheless, other discrepancies arise : Hailer et al. stated that no evidence of on going gene flow was found between polar bears and brown bears, whereas the coalescent model used by Miller et al. yielded that the time when this gene flow stopped was not significantly different from zero. Following the Science article, a comment arose relating two very recent cases of documented hybridization of polar/brown bears in the wild, among them a second generation hybrid. Interestingly, both crosses involved a polar bear female with a brown bear male: thus no cross leading to the introgression of brown bear mtDNA onto polar bear populations has yet been described.
Besides, where Hailer et al. found relatively few shared nuclear data between polar and brown bears, a PCA analysis of the SNPs identified in the ABC, non-ABC and polar bear genomes yielded that 5.5% of one of the ABC genome and 9.4% of the other one are related to the polar bear genome (Fig. 4).
 |
| Fig. 4: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
|
PCA plot of SNP data for ABC1 & 2, polar and non-ABC brown bear (GRZ)
Following this PCA analysis, it is interesting to focus more precisely on the differentiation of populations of polar and brown bears, as the ABC and GRZ seem pretty much apart on the second component axis. Thus Miller et al. arbitrarily chose a subset of 100 SNPs identified from the genomes of all polar bear individuals and resequenced them for 118 individuals (58 polar bears, 9 ABC bears, 51 non-ABC brown bears). The PCA analysis yielded the following plot (Fig. 5).
 |
| Fig. 5: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
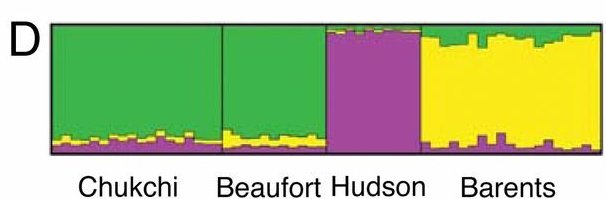
On the one hand, ABC and brown bears cluster together even if we can still discriminate them into two groups. On the other hand, polar bear populations seem much more genetically heterogenous than their sister species counterparts. However one must always remain careful when drawing conclusion on such a low amount of data (100 SNPs). Focusing on the polar populations, the authors performed a structured analysis upon this data (Fig. 6).
 |
| Fig. 5: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
|
|
Structure analysis of 58 polar bear individuals grouped into 4 population
The number of genetic population was set to 3
Here again lies a very striking difference between the two papers. Whereas Miller et al. clearly identified genetic structuring between the populations of polar bears, Hailer et al. used the same type of analysis upon the nuclear variation of their 45 individuals and it led them to conclude that the polar bears were much more genetically homogeneous than the brown bears.
Given the respective data set of both papers, only Miller et al. were able to address the point of adaptation to extreme environment. To do so, they aligned their deeply sequenced genome to the dog genome, choice resulting from a compromise between evolutionary distance and quality of the annotation (as the panda genome has been fully sequenced but being of less good quality). Having thus preserved sinteny accross the bear genomes, they were able to carry admixture analysis for the two ABC genomes (Fig. 6).
 |
| Fig. 6: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
Admixture map of the ABC 1 & 2 diploid genomes region homologous to dog chromosome 11
Blue: polar bear origin, red: brown bear origin
In this particular example, based on the annotation of the dog genome, the authors focus on a gene (ALDH7A1) involved in salt resistance. It appears that copies of this gene in the two ABC bears come from the polar bear. As ABC bears live in a marine environment, the idea hinted behind this plot is that during the hybridization event between polar bear and ABC bears, polar bear (being already adapted to salty environment) copies of this gene introgressed into the ABC population and were subsequently selected for, thus appearing in modern ABC individuals.
Then, using Fst values, they were able to identify a few other genes that might have been selected for during the evolution of polar bears, such as DAG1 (involved in the muscular dystrophy) or BTN1A1 (involved in milk producing).
I think that to address the subject of adaptation in polar bear, a study of positive selection in protein-coding gene is lacking. As authors already conducted transcriptome sequencing of polar and brown bears, annotating gene in their genome, selecting orthologous genes together with other copies from completely sequenced genomes, as dog, panda and other mammals, and then using a model to test for positive selection such as implemented in PAML would be an efficient way to identify genes of interest in the polar (or ABC) bears. Nevertheless, I am very well aware of the tremendous amount of work already performed in this PNAS paper.
Regarding the evolution of the population size in bears, Miller et al. used a pairwise sequentially markovian coalescent model (that uses the length of homozygoteous regions of a diploid genome) to reconstruct the effective population size (number of individual in a perfectly panmictic population leading to the same genetic diversity as our observed population) from the four bear genomes (Fig. 7).
 |
| Fig. 6: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
We observe the very closely related trends of both brown bear genomes and the continuous decline of non polar bears during the Early Pleistocene cooling. Conversly, the population of polar bears increased during this period but seemed very sensitive to the following warming period. Two points were raised when discussing this graph:
- The bump in the polar bear curve signified as the “Post Eemian increase” was not significant when looking at the 95% interval range in the supplementary material
- Knowing from the previous part of the article the extended hybridization between ABC and polar bears, would not the diversity introduced during those event affect the effective population size reconstruction ?
Putting those two papers in parallel allowed us to realize the difficulties of putting in agreement data from various origin, as in this case nuclear, mitochondrial, palaeontological and ecological. The amount of data needed to reconstruct the whole evolutionary history of such a complicated case becomes striking in the light of the work already performed here.
Hailer F, Kutschera VE, Hallström BM, Klassert D, Fain SR, Leonard JA, Arnason U, & Janke A (2012). Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science (New York, N.Y.), 336 (6079), 344-347 PMID: 22517859
Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, Kim HL, Burhans RC, Drautz DI, Wittekindt NE, Tomsho LP, Ibarra-Laclette E, Herrera-Estrella L, Peacock E, Farley S, Sage GK, Rode K, Obbard M, Montiel R, Bachmann L, Ingólfsson O, Aars J, Mailund T, Wiig O, Talbot SL, & Lindqvist C (2012). Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences of the United States of America, 109 (36) PMID: 22826254